Source: Harvard Medical School Aug 28, 2018 7 years, 10 months, 5 days, 13 hours, 21 minutes ago
For decades researchers have worked to shed light on the causes of neurodegenerative disorders, a group of devastating conditions, including Alzheimer's and Parkinson's, that involve the progressive loss of neurons and nervous system function. In recent years, numerous factors, from genetic mutations to viral infections, have been found to contribute to the development of these diseases.
Yet age remains the primary risk factor for almost all neurodegenerative disorders. A precise understanding of the links between aging and neurodegeneration has remained elusive, but research from Harvard Medical School now provides new clues.
In a study published in
Cell on Aug. 23, the research team describes the discovery of a molecular link between aging and a major genetic cause of both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), two related neurodegenerative diseases with shared genetic risk factors.
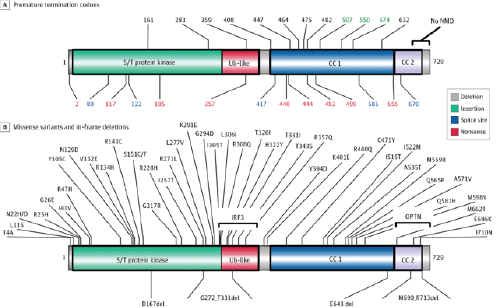
Genetic Variants of TBK1 Found in Patients With Amyotrophic Lateral Sclerosis or Frontotemporal Dementia Mapped on
The findings, the researchers said, reveal possible new targets for treatment of these and other neurodegenerative diseases.
"Our study provides the first description of a molecular event that connects aging with neurodegeneration," said senior study author Junying Yuan, the HMS Elizabeth D. Hay Professor of Cell Biology. "These insights are a critical step towards understanding the mechanisms by which aging predisposes individuals to neurodegeneration."
The results also highlight the need for a better understanding of the biology of neurodegenerative diseases in the context of aging.
"Laboratory models of neurodegenerative diseases often have a missing element, and that is the contribution of age," Yuan said. "We have to understand better the process in its totality, not just its isolated components, to better guide clinical trials and improve our chances of finding effective treatments for these devastating diseases."
RIP rescue
Also known as Lou Gehrig's disease, ALS is a progressive, incurable condition marked by the gradual death of motor neurons. It shares some clinical and genetic features with FTD, which is marked by an early and rapid onset of dementia.
Around one in 10 patients with both diseases carry genetic mutations that cause the partial dysfunction of a protein known as TBK1. Previous studies, including by Yuan and colleagues, have shown that TBK1 is involved in a form of programmed cell death and in neuroinflammation, a hallmark of neurodegenerative disorders. How TBK1 contributes to the development of ALS and FTD, however, was unclear.
In the current study, Yuan and colleagues modeled the reduced levels of TBK1 found in ALS and FTD patients by creating mice that had only one functional copy of the gene that produces TBK1. These mice were healthy and similar in appearance to normal mice. In contrast, those that lacked the gene entirely died before birth.
However, the team found that mice without TBK1 could be fully rescued -- surviving birth and becoming healthy adults -- by blocking the activity of RIPK1, another protein known to play a central role in programmed cell death, neuroinflammation and neurodegenerative disease. Further analyses revealed that TBK1 normally functions to inhibit the activity of RIP
K1 during embryonic development.
This discovery prompted the researchers to investigate another protein, called TAK1, previously known to also inhibit RIPK1 function. When they looked at data on TAK1 expression in human brains, the scientists found that TAK1 expression declines significantly with age. In the brains of patients with ALS, TAK1 expression was further reduced compared with the brains of similarly aged people without ALS.
Brake it down
To model the interaction between partial loss of TBK1 and TAK1 with aging, the team created mice that expressed half the usual amount of TBK1. The mice also expressed half the usual amount of TAK1 in their microglia, the immune cells of the brain where TAK1 is normally most active.
With reductions in both TBK1 and TAK1, these mice displayed traits associated with ALS and FTD, including motor deficits, hind limb weakness, anxiety-like behavior in new environments and changes in brain chemistry. The mice had a reduction in the number of neurons in the brain, and increased motor neuron dysfunction and cell death.
When the team inhibited RIPK1 activity independently of TBK1 and TAK1, they observed a reversal in symptoms.
Like a pair of brakes on a bicycle, TAK1 and TBK1 appear to work together to suppress the activity of RIPK1, and even if one brake fails, the other can compensate, the researchers said. But if both begin to fail, RIPK1 activity increases, leading to cell death and neuroinflammation.
This may be why individuals with TBK1 mutations do not develop ALS and FTD until they become older, when TAK1 levels decline with age, Yuan said.
Several clinical trials are underway to test the safety and
efficacy of drugs that block RIPK1 activity in neurodegenerative and chronic inflammatory diseases, and these findings support the rationale for those trials, Yuan added.
"I think the next couple of years will reveal whether RIPK1 inhibitors can help ALS and FTD patients," she said. "I think our study makes us more confident those efforts might work."
Despite numerous large-scale clinical trials, however, no effective therapeutics have yet been developed for neurodegenerative diseases. This research now establishes a model of study that incorporates both aging and genetic risk for ALS and FTD, which may have broad implications.
"Many trials have been launched based on data from studies in mice, but how can a two-year-old mouse, for example, fully reflect what happens in an 80-year-old patient with Alzheimer's?" Yuan said. "We need to develop new thinking on how to model these diseases to incorporate the element of aging, and we think this study is an important step toward that goal."
The scientists are currently investigating why TAK1 levels decline with age and its potential role in other neurodegenerative diseases.
Reference: Daichao Xu, Taijie Jin, Hong Zhu, Hongbo Chen, Dimitry Ofengeim, Chengyu Zou, Lauren Mifflin, Lifeng Pan, Palak Amin, Wanjin Li, Bing Shan, Masanori Gomi Naito, Huyan Meng, Ying Li, Heling Pan, Liviu Aron, Xian Adiconis, Joshua Z. Levin, Bruce A. Yankner, Junying Yuan. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging.
Cell, 2018; DOI: 10.1016/j.cell.2018.07.041