Heart Failures: Scientists Discover That Defective Desmosomal Adhesion Causes Arrhythmogenic Cardiomyopathy, A Heart Condition Affecting Young Adults
Heart Failure - Defective Desmosomal Adhesion Dec 18, 2022 3 years, 6 months, 1 week, 2 days, 12 hours, 18 minutes ago
Heart Failures: A team of international medical researchers have recently discovered that defective desmosomal adhesion causes arrhythmogenic cardiomyopathy, a heart condition affecting many young adults especially athletes.
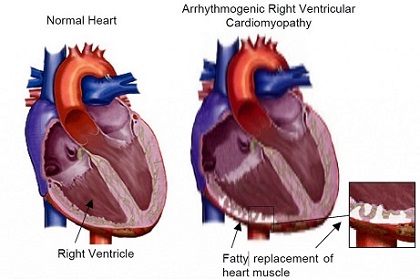
Individuals with arrhythmogenic cardiomyopathy (ACM) typically present with features ranging from impaired cardiac function and ventricular arrhythmia to sudden cardiac death as a result of
Heart Failure.
Although in most cases arrhythmogenic cardiomyopathy (ACM) is a genetic disorder that is thought to affect 1 in 5,000 people, where the heart muscle (myocardium) is replaced by both scar (fibrosis) and fat, affect predominantly the right ventricle, the left ventricle, or both ventricles…..new studies have emerged that exposure to the SARS-CoV-2 virus can also cause arrhythmogenic cardiomyopathy or conditions very similar to it!
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8901242/
https://www.frontiersin.org/articles/10.3389/fcvm.2021.759119/full
https://www.mdpi.com/2308-3425/9/4/96
https://jcmr-online.biomedcentral.com/articles/10.1186/s12968-022-00872-2
https://scholarlycommons.hcahealthcare.com/hcahealthcarejournal/vol1/iss0/9/
https://www.ahajournals.org/doi/full/10.1161/CIRCULATIONAHA.121.058272
https://academic.oup.com/ehjcr/article/5/7/ytab220/6330927
https://www.biorxiv.org/content/10.1101/2020.09.14.296178v1.full
The study team led by researchers from the University of Basel-Switzerland, by genetically modifying mice to develop the similar disease of arrhythmogenic cardiomyopathy was able to uncover previously undiscovered mechanisms behind the condition and also potential treatment targets.
The study team also comprised of scientists from University Hospital Basel-Switzerland, University of California San Diego-USA, University Hospital Frankfurt, Goethe University-Germany and University Hospital, LMU Munich-Germany.
Of late, we have been witnessing increased reports of young celebrities and sports stars dying from sudden cardiac arrest especially males.
Dr Volker Spindler, anatomist and head of the Cell Adhesion group at the University of Basel’s Department of Biomedicine told Thailand
Medical News, &
amp;ldquo;Arrhythmogenic cardiomyopathy leads to arrhythmia with a loss of cardiac muscle cells, deposits of connective tissue, and fat within the cardiac muscle. This can cause sudden cardiac death, often during exercise or any strenuous activity.”
At present, it is recognized that a number of gene mutations can trigger the condition. There is no treatment, even with an early diagnosis; only symptom management options are available.
Cardiologist Dr Gabriela Kuster, who heads the Myocardial Research group at the Department of Biomedicine at the University of Basel added, “Patients are advised to avoid any competitive or endurance sports and have to take medications such as beta-blockers. Where appropriate, a catheter ablation may be performed or an implantable defibrillator may be used” Sometimes the only option is a heart transplant.”
For the study team, the starting point for the research was the notion that many of the mutations affect structures known as the desmosomes.
Desmosomes are protein clusters on the surface of cardiac muscle cells that ensure a tight connection between the cells.
First author, Dr Camilla Schinner from the University of Basel added, “You can imagine these clusters to act like a piece of Velcro. This led to the theory that the mutations reduce adhesion between the cells, thus weakening the cardiac muscle.”
In order to test this hypothesis, the study team introduced a mutation similar to that found in patients into the genome of mice. The cardiac function of these animals was then examined by. The study findings showed the genetically modified animals showed a heart disease with arrhythmia that resembled arrhythmogenic cardiomyopathy in humans.
Importantly, microscopic and biochemical analysis showed reduced adhesion between the cardiac muscle cells. The study team also observed the scarring of the cardiac muscle typical for this disease.
The study team next explored how diseased cardiac muscle differed from healthy conditions at the molecular level.
Interestingly, it was found that mice with the mutation showed an increased amount of a particular protein at the Velcro-like structures of the heart muscle cells. This leads via a series of events, to connective tissue deposition and scarring of the heart.
The addition of a substance that blocks this cascade prevented disease progression. The study team sees a potential new treatment approach from this finding.
The study findings were published in the peer reviewed journal: Circulation (A Journal of the American Heart Association)
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.121.057329
Typically, arrhythmogenic cardiomyopathy (ACM) is characterized by progressive loss of cardiomyocytes with fibrofatty tissue replacement, systolic dysfunction, and life-threatening arrhythmias.
A substantial proportion of ACM is caused by mutations in genes of the desmosomal cell–cell adhesion complex, but the underlying mechanisms are not well understood.
The study team investigated the relevance of defective desmosomal adhesion for ACM development and progression.
The researchers mutated the binding site of DSG2 (desmoglein-2), a crucial desmosomal adhesion molecule in cardiomyocytes. This DSG2-W2A mutation abrogates the tryptophan swap, a central interaction mechanism of DSG2 on the basis of structural data.
Impaired adhesive function of DSG2-W2A was confirmed by cell–cell dissociation assays and force spectroscopy measurements by atomic force microscopy.
The DSG2-W2A knock-in mouse model was analyzed by echocardiography, ECG, and histologic and biomolecular techniques including RNA sequencing and transmission electron and super-resolution microscopy.
The findings were compared with ACM patient samples, and their relevance was confirmed in vivo and in cardiac slice cultures by inhibitor studies applying the small molecule EMD527040 or an inhibitory integrin-αVβ6 antibody.
The study findings showed that the DSG2-W2A mutation impaired binding on molecular level and compromised intercellular adhesive function.
It was found that mice bearing this mutation develop a severe cardiac phenotype recalling the characteristics of ACM, including cardiac fibrosis, impaired systolic function, and arrhythmia.
A detailed comparison of the transcriptome of mutant mice with ACM patient data suggested deregulated integrin-αVβ6 and subsequent transforming growth factor–β signaling as driver of cardiac fibrosis.
Importantly, blocking integrin-αVβ6 led to reduced expression of profibrotic markers and reduced fibrosis formation in mutant animals in vivo.
The study findings showed that disruption of desmosomal adhesion is sufficient to induce a phenotype that fulfils the clinical criteria to establish the diagnosis of ACM, confirming the dysfunctional adhesion hypothesis.
Deregulation of integrin-αVβ6 and transforming growth factor–β signaling was identified as a central step toward fibrosis. A pilot in vivo drug test revealed this pathway as a promising target to ameliorate fibrosis. This highlights the value of this model to discern mechanisms of cardiac fibrosis and to identify and test novel treatment options for ACM.
The study team are pursuing further studies including clinical trails using existing drugs to target blocking integrin-αVβ6.
For the latest on
Heart Failure, keep on logging to Thailand Medical News.


 Share
Share
 Tweet
Tweet
 Share
Share