

BREAKING NEWS
For the latest on Thailand Medical Industry, Thailand Doctors, Thailand Medical Research, Thailand Hospitals, Thailand Wellness Initiatives and the latest Medical News
World Sickle Cell Day is June 19 and was created by the United Nations in 2006 to raise global awareness for sickle cell disease as a public health problem. Sickle cell disease is an extremely debilitating medical condition. It is characterized by chronic pain with frequent bouts of acute severe pain that requires large amounts of narcotics that often provide incomplete relief. It is also marked by social stigma that makes others, including medical professionals, view sickle cell patients as “drug seekers” due to their need for large amounts of pain medication.
Sickle cell disease is a hereditary disease that affects a gene that controls our blood cells. A small change (called point mutation) in this particular gene causes deformities of the red blood cells. Having one of these affected genes will lead to a condition called sickle cell trait or carrier state. This condition can be passed to one’s offspring with minor effects to the carrier. However, having a pair of this sickle-affected genes or one sickle-affected gene paired with another defective gene of another type can lead to severely deformed red blood cells. With the deformity, these cells become rigid and sticky. This results in blockage of small blood vessels as seen in Figure 1.
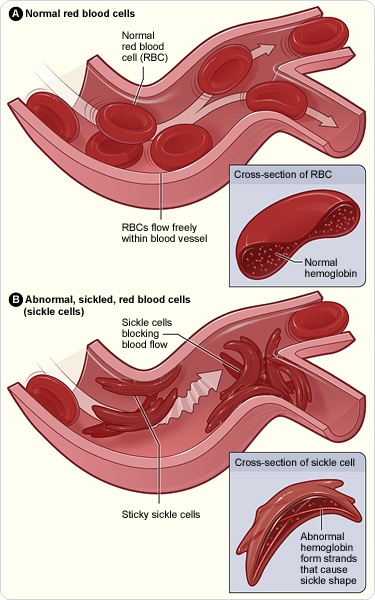
When the blood vessels are blocked, oxygen and nutrients cannot be carried to the area covered by the affected blood vessels and tissue in the area will die and cause severe inflammation. It’s as though small heart attacks are occurring throughout the body, causing severe pain in sickle cell patients. This happens not just once or twice, but closer to several times a year.
Typical sickle cell patients experience chronic underlying pain that they have to live with throughout their lives. They have an annual average of three episodes of severe pain that requires hospitalization and narcotics. Unfortunately, many people—including medical care providers—do not understand their pain and under treat them or simply ignore treatment and label them as “drug seekers.” Sickle cell patients’ pain is real and is triggered by damage to major parts of their bodies, including the heart, lungs, kidneys and brain. Also, they are very susceptible to infection, which can be life threatening. These organ damage-induced bouts of pain and infectious episodes are two causes of mortality at a very young age. In developed countries, the average life span of sickle cell patients has increased to about 40 years due to better access to medical care; however, in underdeveloped countries, 90 percent will not survive past age five.
Worldwide, it is believed that there are in excess of four million sickle cell disease patients today. The majority of them are in Africa, mostly in the Central and Western regions. About 100,000 patients, primarily of African or Latin origin, live in the United States. Another 100,000 are in Europe. The disease also affects four to five million people in the Middle East, India, South America and Caribbean.
This hereditary condition is believed to have started in the Western and Central parts of Africa. In those areas, sickle cell trait or carrier status was beneficial because the single gene actually protected a person from malaria. As a result, those with the gene survived the malaria epidemic in the affected regions of Africa. However, people who inherited the affected gene from both parents, had the condition called sickle cell anemia, which meant they were likely suffer and die from the disease.
Aside from sickle cell anemia, other genetic changes can result in similar conditions and are categorized under sickle cell disease. Sickle cell anemia is the most common type of the sickle cell disease. Other major conditions in the same category include: SC disease (a combination of sickle mutation and hemoglobin C mutation) and Sβ-thalassemia (a combination of sickle mutation and β-thalassemia mutation). In all of these conditions, patients can have severe painful episodes and organ damage.
One of the first manifestations of symptoms can occur as early as 6 months of age. A baby with sickle cell disease can have blockage of blood vessels in his or her hands and feet, which is referred to as hand-foot syndrome with severe swelling of these parts of the body. The baby will cry profusely due to the pain.
During childhood, splenic sequestration syndrome can be seen. In this condition, sticky blood vessels suddenly stop up the small vessels of the spleen. A spleen is like a rubber bag that can stretch. Therefore when the outlet of this organ is blocked it becomes a dam that can hold two-thirds of the body’s blood volume. As the spleen stretches out, it can retain most of the blood that is otherwise needed in circulation. This is a life-threatening condition.
Infection is very common throughout lifetime of sickle cell patients. It is especially true in childhood when children are building up their immune systems. These children have several hundred times higher chance of contracting a life threatening infection such as meningitis, pneumonia and sepsis. As a preventative measure in developed countries, it is customary for these children to take prophylactic antibiotics until about five years old. This has significantly decreased the incidence of infection and childhood mortality.
Another common complication is stroke. During childhood, the common type is due to occlusions of large blood vessels in the brain. As the patients get older, a more common type is due to breakage of the weakened blood vessels in the brain, resulting in hemorrhage.
Chest syndrome is a rapidly progressive condition of lungs and lung coverings. It may start out as pain in the chest but within hours it can shut down the lungs. This is something that can be mistaken for muscle ache or mild pleurisy at first unless a care provider is aware of it and evaluates for it early. Obviously, this condition requires rapid intervention before it leads to respiratory failure.
Over the years, due to the need for blood transfusion and the tendency of sickle cell disease to accumulate iron, iron overload can also affect older patients. Iron overload can cause organ damage on its own. When combined with sickle cell disease, the patient will have very little chance in preserving their organs.
These are only a few of the complications this disease can cause. Sickle cell disease patients can also experience irregular growth of bones, priapism, pregnancy complications and more.
Today, we have limited measures for treating sickle cell disease. The use of antibiotics over the last 50 years has done much to prevent deadly infection among children with the disease, leading to major improvements in survival rates in developed countries. Another example of progress is the development of the “Doppler device,” which can determine which children are at risk for a major stroke. By placing this device over the middle cerebral artery, the blood flow speed can be detected. Faster blood flow suggests a narrowed artery. The narrower the artery, the higher the chance of its closure. Thus, a patient who has blood flow faster than 200cm/sec over the middle cerebral artery can receive blood transfusions on regular basis as a preventive measure. The Doppler device has led to a decreased incidence of strokes in children with sickle cell disease.
The only known cure for sickle cell disease is stem cell transplantation; however, this is available to a very limited number of patients due to scarcity of matched donors. In most cases, the donors are siblings. There is a one in four chance that a sibling could be a match. There is also a one in four chance that a sibling also has sickle cell disease. Outside of sibling donors, the chance of matching is incredibly rare—this includes parents and other relatives. This treatment is also difficult to complete due to up to a 5% chance of death from the treatment itself and related complications.
Since the mid 90’s, an oral medication called Hydroxyurea has been available for prevention of “painful crises.” For those who can take the medication on a regular basis, it has improved their quality life immensely. However, it is a medicine that was originally designed for certain types of cancer and has many side effects, including suppression of white blood cells that fight infection, skin ulcers, stomach problems and anemia. It can also cause birth defects. While it is much less toxic than a typical anti-cancer agent, Hydroxyurea is not considered one of the safest medications. The statistics indicate that only about 1.5% of sickle cell patients regularly take this agent.
There are other treatments on the horizon. Some of the medications are being studied to reduce the severity and duration of pain during “painful crises.” Others are designed to prevent these terrible episodes of pain and other symptoms. One medication that my company is working on, in the last phase of FDA supervised clinical trial (phase III), uses L-glutamine. During the phase II study, daily use of the medication led to more than a 50% drop in the episodes of “painful crises” and nearly a 40% drop in hospitalization episodes. Additionally, there were no reports of major side effects.
Gene therapy is being studied as a future treatment option. There are genes that may actually improve or cure the condition. The issue now is determining how to integrate these genes into patients’ cells. Once this gene-carrying vehicle is discovered, gene therapy will be a reality.
Many capable investigators, with the help of the patients, are devising solutions for this disease. Someday, sickle cell disease may be seen as a “pernicious anemia,” which once was a deadly disease but, thanks to proper treatment, now is a curable condition.

Dr. Yutaka Niihara was born in Tokyo, Japan and immigrated to Hawaii in 1973 at age 13. He studied Religion at Loma Linda University receiving his BA and later received his MD from the same institution. Dr. Niihara received his Internal Medicine training at Kettering Medical Center in Kettering, Ohio and his Hematology/Oncology training at Harbor-UCLA Medical Center. He received his MPH from Harvard School of Public Health.
Dr. Niihara joined as Faculty of David Geffen School of Medicine in 1992 and currently serves as Clinical Professor of Medicine. He has focused his research work on sickle cell disease since 1992. His honors include: Recipient of NIH FIRST award in 1997 and of FDA Orphan Drug Development Award in 2000. Dr. Niihara founded Emmaus Medical, Inc. in 2003 as a vehicle for commercial development of the treatment for sickle cell disease, which was started at LA Biomed at Harbor-UCLA Medical Center.