BREAKING! German Researchers Discover That SARS-CoV-2 Virus Proteins Manipulate Autophagy In Human Host Cells
Source: SARS-CoV-2-Research Aug 02, 2021 4 years, 11 months, 1 day, 18 hours, 42 minutes ago
Since the debut of the SARS-CoV-2 coronavirus in Wuhan-China in late 2019, scientists are constantly discovering new characteristics and facets of the complex novel coronavirus and also its pathogenesis and ways its affects various aspects of the cellular pathways of the human host.
.jpg)
In a new study by researchers from the Institute of Molecular Virology, Ulm University Medical Center-Germany, it was found that the SARS-CoV-2 coronavirus was able to manipulate autophagy in the human host.
A key and critical component of the innate immune defenses, macroautophagy/autophagy targets viruses and viral components for lysosomal degradation and exposes pathogen-associated molecular patterns to facilitate recognition.
However, it has been found that certain viruses evolved sophisticated strategies to antagonize
autophagy and even exploit it to promote their replication.
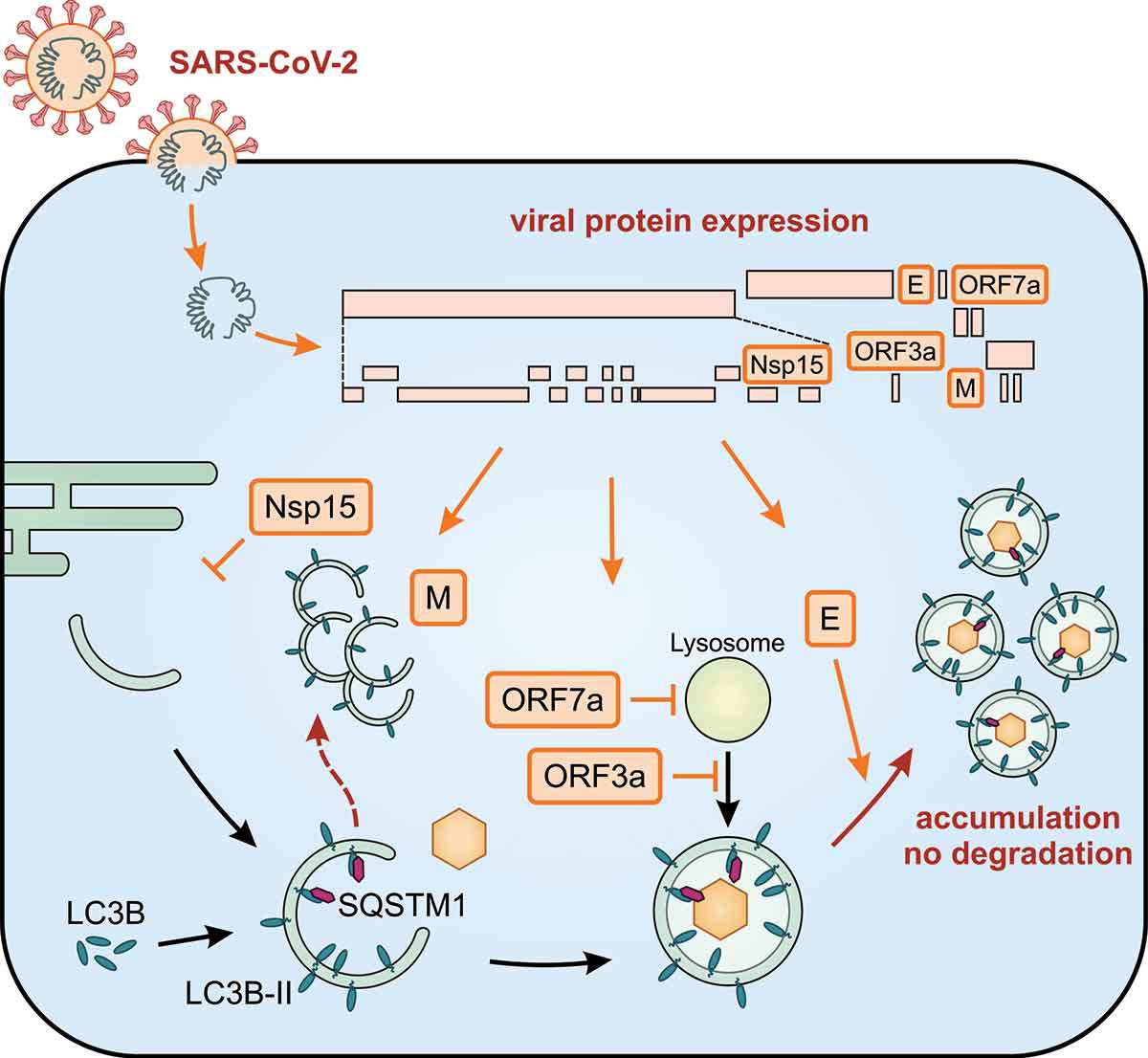 The impact of SARS-CoV-2 proteins on autophagy
After nucleation, the autophagosome is elongated and decorated with LC3B-II. SQSTM1 is an autophagy receptor that recruits cargo. Infection of cells by SARS-CoV-2 leads to the expression of viral proteins (orange boxes) and autophagy is substantially regulated by Nsp15, M, ORF7a, ORF3a, and E. Altogether, infection with SARS-CoV-2 leads to an accumulation of autophagosomes and prevents degradation of their cargo.
The impact of SARS-CoV-2 proteins on autophagy
After nucleation, the autophagosome is elongated and decorated with LC3B-II. SQSTM1 is an autophagy receptor that recruits cargo. Infection of cells by SARS-CoV-2 leads to the expression of viral proteins (orange boxes) and autophagy is substantially regulated by Nsp15, M, ORF7a, ORF3a, and E. Altogether, infection with SARS-CoV-2 leads to an accumulation of autophagosomes and prevents degradation of their cargo.
The German study team systematically analyzed the impact of individual SARS-CoV-2 proteins on autophagy.
The study findings showed that E, M, ORF3a, and ORF7a cause an accumulation of autophagosomes, whereas Nsp15 prevents the efficient formation of autophagosomes. Consequently, autophagic degradation of SQSTM1/p62 is decreased in the presence of E, ORF3a, ORF7a, and Nsp15. Notably, M does not alter SQSTM1 protein levels and colocalizes with accumulations of LC3B-positive membranes not resembling vesicles. Infection with SARS-CoV-2 prevents SQSTM1 degradation and increases lipidation of LC3B, indicating overall that the infection causes a reduction of autophagic flux.
The detailed study findings showed that the accessory proteins ORF3a and ORF7a both block autophagic degradation but use different strategies. While ORF3a prevents the fusion between autophagosomes and lysosomes, ORF7a reduces the acidity of lysosomes.
The study findings found that Nsp15, E, M, ORF3a, and ORF7a of SARS-CoV-2 manipulate cellular autophagy, and the study also determined the molecular mechanisms of ORF3a and ORF7a.
The study findings were published in the peer reviewed journal: Autophagy.
https://www.tandfonline.com/doi/full/10.1080/15548627.2021.1953847
The current COVID-19 pandemic has claimed more than 4.23 million lives worldwide and is caused by a novel single-stranded RNA virus – t
he SARS-CoV-2 coronavirus. The rapid spread of this virus is attributed to its high infectiousness and its ability to escape and counteract host immune responses, including the newly discovered autophagy manipulation.
Autophagy is the natural, conserved degradation lysosome-dependent regulated mechanism of the cell that removes unnecessary or dysfunctional components. It allows the orderly degradation and recycling of cellular components. It plays a vital role in the antiviral defenses of the host. It targets a virus or viral components for lysosomal degradation.
However several viruses have developed means to evade the lysosome-dependent degradation by autophagy. These viruses sometimes evolve to manipulate the autophagic machinery such that no inhibition is encountered during viral replication, membrane trafficking, and fusion processes.
Past research has revealed that infection of human cells with SARS-CoV-2 decreases autophagic flux. This can be evaluated by studying two markers, i.e., the increased presence of the processed form of LC3B and LC3B-II and an accumulation of SQSTM1.
https://www.sciencedirect.com/science/article/pii/S2211124721004654
Interestingly in vitro studies revealed that the virus was not greatly affected by the initiation of autophagy using rapamycin, but remained sensitive to innate immune stimulation via interferons. This experiment showed that SARS-CoV-2 could efficiently evade the antiviral functions of autophagy.
The study team mapped out the molecular mechanism behind the evasion of antiviral functions of autophagy by the SARS-CoV-2 coronavirus and provided a systematic analysis of the impacts of SARS-CoV-2 proteins on autophagy.
The study team analyzed the impact of 29 out of the 30 known SARS-CoV-2 proteins on autophagy. They found a decrease in the number of LC3B-positive autophagosomes in the presence of Nsp15.
The team conducted a Western Blotting analysis and found that Nsp15 affects the MTOR axis. Additionally, the expression of viral proteins such as envelope (E), membrane (M), ORF3a, and ORF7a resulted in a robust accumulation of membrane-associated LC3B.
The German study team re-examined the role of the proteins on autophagosome numbers to understand if the accumulation of LC3B is associated with de novo induction of autophagy or rather caused by a block of autophagic flux.
Importantly the activation of autophagy by rapamycin showed no impact on E, M, ORF3a, and ORF7a. Further, bafilomycin A1 masks their effects. The results indicate that these viral proteins prevent autophagic flux. Similarly, the expression of E, ORF3a, ORF7a, and Nsp15 causes the accumulation of SQSTM1.
The study team also reported that even though M protein triggers LC3B processing and increases LC3B-membrane localization, it does not restrict SQSTM1 degradation.
This study finding indicates that this viral protein does not inhibit classical autophagy. The study also found SARS-CoV-2 ORF3a, ORF7a, M, and Nsp15 in autophagy to be a conserved sequence. When comparing sequences, the closest similarity was found with the conserved sequences of bat coronavirus RaTG13 and SARS-CoV-1.
The study team also conducted a proteomic analysis to assess the molecular mechanisms by which ORF3a and ORF7a block autophagic flux.
The study team found that both ORF3a and ORF7a colocalize with late endosomal markers and trans-Golgi network markers. However, these do not localize with early endosomal markers. The pH-sensitive molecular probes were used to study the maturation of autophagosomes and with the help of the mCherry-GFP-LC3B reporter system, researchers found that both ORF3a and ORF7a prevent the acidification of autophagosomes. This study has further reported that ORF7a lowers the number of acidic lysosomes, whilst ORF3a enhances their accumulation.
The study findings indicate that ORF7a lowers lysosome acidity and promotes autophagosomal degradation, whereas ORF3a blocks fusion between lysosomes and autophagosomes.
A significant finding of the current study is that ORF7a-mediated deacidification of lysosomes may lead to the release of virions by avoiding degradation. Additionally, the viral E protein might be responsible for redirecting autophagosomes to assist SARS-CoV-2 virion release through the lysosomal route.
The study team concluded that the viral proteins are used by SARS-CoV-2 to manipulate autophagy. The study findings have also highlighted the molecular mechanisms of ORF3a and ORF7a in autophagic. These results help in understanding the pathogenesis of SARS-CoV-2 and the mechanisms via which they evade autophagy induction. This research has also paved the way for future studies to understand how E, M, and Nsp15 modulate autophagy.
For the latest
SARS-CoV-2-Research, keep on logging to Thailand Medical News.