Source: Thailand Medical News Nov 28, 2019 6 years, 7 months, 3 days, 2 hours, 52 minutes ago
Typically, It is the wish of every molecular
geneticist: an easy-to-use program that compares data sets from different cellular conditions, identifies enhancer regions and then assigns them to their target
genes. A research team led by Martin Vingron at the Max Planck Institute for Molecular
Genetics in Berlin has now developed a program that masters all of this.
Dr Martin Vingron, Director and Head of the Department of Bioinformatics at the Max Planck Institute for Molecular
Genetics in Berlin told
Thailand Medical News, "DNA is pretty boring, since it is practically the same in every cell. When the
genome is like the book of life, I am most interested in the side notes.”
These "notes" are small chemical marks attached to the DNA molecule that do not alter the
genetic information itself, but influence what happens to the DNA at the respective site. In other words, these marks have an
epigenetic effect. They serve as regulators of
genomic regions that are responsible for the activation and deactivation of
genes, such as promoters and enhancers.
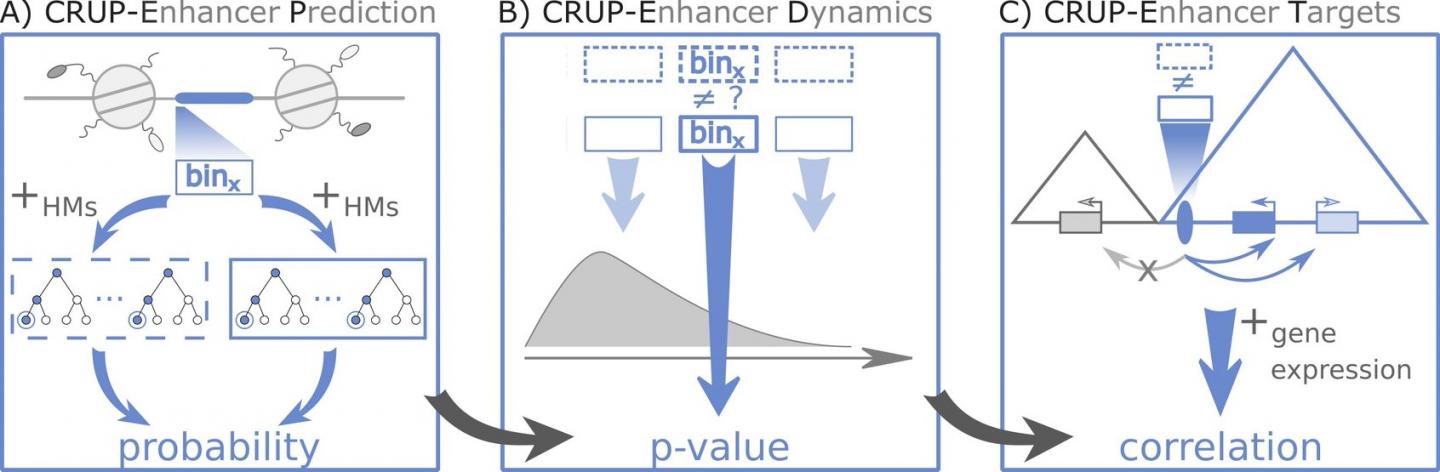 Three steps to complete enhancer analysis with CRUP: First, an algorithm identifies the active enhancers. Then, it reveals epigenetic differences between data sets and at last, assigns differential enhancers to their respective target genes. Credit: C-BY 4.0, Ramisch A
Three steps to complete enhancer analysis with CRUP: First, an algorithm identifies the active enhancers. Then, it reveals epigenetic differences between data sets and at last, assigns differential enhancers to their respective target genes. Credit: C-BY 4.0, Ramisch A
In numerous complex diseases, the
epigenetic control of
genes does not work correctly and is of great interest for scientists. The analysis of these regions in the lab, however, is often cumbersome, time-consuming and complicated. That's why Vingron and his team decided to develop a new program package called Condition-specific Regulatory Units Prediction (
CRUP) which simplifies analysis and solves several practical problems.
Bioinformatician Verena Heinrich who developed the package commented, "We wanted to combine the common steps in the process of enhancer prediction in a simple, universal program."
CRUP simplifies the analysis in many regards. The machine learning algorithm is not limited to specific cell- or tissue-types. It does not need to be recalibrated prior to each analysis of a data set and allows comparative study of several data series. The tool, which was developed by Heinrich and doctoral student Anna Ramisch, is still easy to use.
CRUP specifically identifies and characterizes enhancers, DNA segments that stimulate or "enhance" the transcription of genes. These regions attract proteins that attach to promoter sequences which function as a switch for each
gene. However, which enhancer controls the right
genes at correct time often remains a mystery. "Enhancers and their associated
genes can be located far away from each other," says Heinr
ich. "This makes it difficult for us to assign the regulatory sequences to their respective targets."
Typically. the
genome contains hundreds of thousands of enhancers which are active in different phases in the life of a cell like during growth, maintenance, or disease. When the DNA is tightly packed like a wool thread on spools of carrier proteins called histones, the regulatory sequences are in a "resting" state. They only become active by chemical modifications to the histone proteins. Then, sections unwrap from the DNA clusters, get exposed, and become accessible to molecules that activate
genes. The analysis of histone proteins by
chromatin immunoprecipitation (
ChIP) in tandem with DNA sequencing then reveals which enhancers are active and which are not.
These critical
ChIP data are the input for the newly developed program.
CRUP first examines all sequences and then decides whether it is an enhancer or not. The classification algorithm is based on artificial intelligence, which is trained with information from mouse embryonic stem cells. It detects enhancer regions in many other animal species or tissues, as Heinrich and her colleagues demonstrated on a diverse set of data provided by the German
Epigenome Program (DEEP).
In the next step,
CRUP can be fed multiple data sets and the program finds where they differ. This makes it possible to interpret a series of measurements or pinpoint differences between tissues.
Epigenetic changes to enhancers become apparent over time, or when comparing healthy and diseased tissues. The third and final step of the analysis is the mapping of
genes to their respective enhancers. "We asked: What part of the
genome is active at the same time in the same place?" explains Heinrich. To achieve this,
CRUP links the enhancer analysis with transcription data that reveal which
genes are active, and experiments that tell which parts of the DNA strand are close to each other.
The researchers tested their program in a practical setting. They analyzed the tissue of mice with the immune disease rheumatoid arthritis and compared it with data from healthy animals.
CRUP identified more than 200 differences in enhancer regions, some of which had already been associated with the disease in other studies. The
genes that
CRUP assigned to these enhancers have also been shown to play a role in disease.
Dr Martin Vingron concluded, "Our program reliably identifies candidates for disease-associated enhancers and links them to their target
gene."
The team hopes the new tool will make the field more accessible as well as accelerate research to help identify the causes of complex human diseases. "
CRUP should be particularly useful for all the research groups that do not have a team of bioinformaticians at hand."
Reference: Ramisch et al. 2019. CRUP: a comprehensive framework to predict condition-specific regulatory units. Genome Biology. DOI: https://doi.org/10.1186/s13059-019-1860-7.